| |
Editor's
note: A combination of three recent developments makes homocysteine
measurement and its clinical utility timely topics.
First,
as part of DPC's commitment to its customers and their testing needs,
the Company recently acquired a homocysteine assay pass-through license
from Competitive Technologies, Inc. (CTT). The license permits DPC customers
in the US to continue to perform homocysteine assays without concern about
possible infringement of the CTT homocysteine assay patent.
DPC
homocysteine assays are intended for the quantitative determination of
L-homocysteine in human plasma or serum. They can assist in the diagnosis
and treatment of patients suspected of having hyperhomocysteinemia or
homocystinuria. In the US, homocysteine is regarded as a marker of cardiovascular
disease (CVD) risk. In Europe, the interest in homocysteine as an indicator
of vitamin deficiency surpasses interest in its utility as a marker for
CVD.
Second,
DPC's IMMULITE® 2500 Homocysteine assay is now available worldwide. This
fully automated, random access method will allow for consolidation on
a single platform of testing with other assays such as Vitamin B12 and
Folic Acid when they become available. DPC also offers homocysteine assays
on the IMMULITE®, IMMULITE® 1000 and IMMULITE® 2000 platforms.
Third,
a consensus document* of the German, Austrian and Swiss Homocysteine Association
(D.A.CH.-Liga Homocystein) spells out targeted populations for testing
and recommended homocysteine cutoffs for treatment. More about homocysteine
testing can be found on DPC's website at www.dpcweb.com/homocysteine/index.html.
It
is in this context that we are pleased to present again an article on
homocysteine by Dr. Donald Jacobsen. The article provides an overview
of homocysteine metabolism and the various conditions in which it may
figure as a risk factor and even a participant in pathological processes.
Homocysteine
is receiving a lot of attention these days as a new risk factor for a
variety of diseases. For more than a decade, the vast majority of nearly
100 case-control retrospective and prospective studies have shown that
homocysteine is a strong independent risk factor for coronary artery disease,
cerebrovascular disease and peripheral vascular disease. More recent studies
now implicate homocysteine as a risk factor for neural tube defects in
newborns and for cognitive dysfunction disorders such as vascular dementia
and Alzheimer's disease.
What
is homocysteine, where does it come from, and how is it metabolized?
Homocysteine is a normal metabolite of the essential amino acid methionine
(Figure 1). Structurally, it closely resembles methionine and cysteine;
all three amino acids contain sulfur. They are metabolically linked to
each other as shown in Figure 2. Since foods contain little or no free
homocysteine, nearly all of the homocysteine in the body is derived from
methionine in animal and plant proteins.
Homocysteine
metabolism is driven by several B-complex cofactors. Folate and vitamins
B2, B6 and B12 are used in the remethylation
pathway; and vitamin B6 is used in the transsulfuration pathway
(Figure 2). Deficiencies of folate, vitamin B6 or vitamin B12
can lead to impaired homocysteine metabolism and hyperhomocysteinemia.
In addition, mutations in the genes coding for methylenetetrahydrofolate
reductase (MTHFR), methionine synthase (MS) and cystathionine b-synthase
(CBS) may also produce hyperhomocysteinemia. Subjects who inherit two
identical defective alleles may have little or no enzyme activity (e.g.,
for CBS). This can result in severe hyperhomocysteinemia and the rare
disease known as homocystinuria. Without treatment, most affected individuals
will experience a cardiovascular event before age 30.
The
determinants of mild hyperhomocysteinemia, commonly seen in patients with
cardiovascular disease, are multifactorial and involve both genetic and
acquired components. Gene-nutrient interactions such as homozygosity for
thermolabile MTHFR, and low-folate nutritional status, can result in mild
hyperhomocysteinemia. Approximately 12 percent of the Caucasian population
is homozygous for thermolabile MTHFR. Smoking, excessive coffee consumption
and lack of exercise are associated with elevations in homocysteine as
well.

|
|
|
Figure
1. Structures of methionine, homocysteine and cysteine.
|
|
|
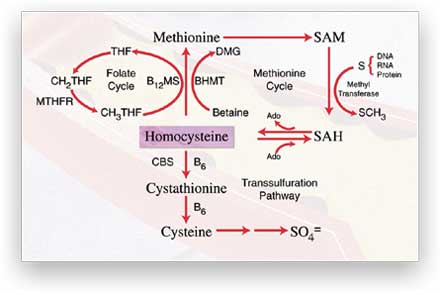
| Figure 2. Major
pathways of homocysteine metabolism in the liver and kidneys. Homocysteine
is generated in a cycle through S-adenosylmethionine (SAM) and S-adenosylhomocysteine
(SAH). Remethylation of homocysteine back to methionine is carried
out by vitamin B12-dependent methionine synthase (B12MS)
and betaine-homocysteine methyltransferase (BHMT). Homocysteine is
also converted to cysteine through the transsulfuration pathway, initiated
by B6-dependent cystathionine b-synthase
(CBS). The folate cycle generates 5-methyltetrahydrofolate (CH3THF)
for the remethylation of homocysteine back to methionine. Other abbreviations:
DMG, dimethylglycine; Ado, adenosine; THF, tetrahydrofolate; CH2THF,
5,10-methylenetetrahydrofolate; SO4 =, sulfate. |
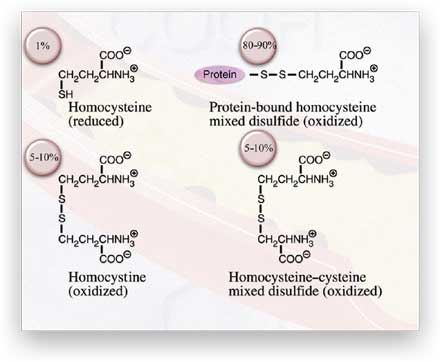
What is hyperhomocysteinemia and how is it determined?
The term can be defined simply as "elevated blood homocysteine" but the
actual situation is more complex. When homocysteine is transported out
of cells into circulation, it reacts with other compounds containing sulfhydryl
(-SH) or disulfide (-S-S-) groups. As a result of these reactions almost
all of the homocysteine in circulation is converted to a disulfide (oxidized)
form. Less than 1 percent of total plasma homocysteine is found as the
free -SH form. The disulfide forms include the symmetrical dimer homocystine
and mixed disulfides with cysteine and cysteine-containing plasma proteins
(Figure 3). In fact, over 70 percent of circulating homocysteine is carried
as a mixed disulfide by plasma proteins.
Sensitive
and reliable assays for plasma total homocysteine (tHcy) were developed
in the mid- to late 1980s. This technical achievement was largely responsible
for establishing homocysteine as a major independent risk factor for cardiovascular
disease. In practice, plasma samples are treated with strong reducing
agents to break disulfide bonds, thus liberating free homocysteine and
other small thiols such as cysteine and glutathione. The thiols are usually
derivatized with a reporter group, separated and detected. Thiol-specific
fluorescent reporter groups are commonly used, and separations are usually
achieved by high-performance liquid chromatography (HPLC), after which
the compounds are detected fluorometrically (HPLC-FD). Other methods use
HPLC with electrochemical detection (HPLC-ED), or gas chromatography with
mass spectrometry (GC/MS). Immunoassays for plasma tHcy were introduced
about ten years ago.
|
|
|
Figure
3. The circulating forms of homocysteine that make up plasma total
homocysteine.
|
What is a "normal" plasma tHcy?
Until recently, the normal range for plasma tHcy was considered to be
5 to 15 µmol/L. It is now widely accepted that the upper limit of normal
may be around 10 µmol/L for middle-aged adults and that risk for cardiovascular
disease occurs if plasma tHcy exceeds this value. However, it is now also
recognized that homocysteine levels increase with age, perhaps as a result
of micronutrient deficiencies due to malabsorption. In the future, it
is likely that age-specific reference ranges will be established. Premenopausal
women have approximately 20 percent lower values than their male counterparts,
suggesting that homocysteine metabolism may be regulated to some extent
by hormones. Patients with coronary artery disease and other cardiovascular
diseases usually have mild hyperhomocysteinemia (>10 to 25 µmol/L) with
an incidence of 30 to 50 percent. Almost all patients with end-stage renal
disease have hyperhomocysteinemia that tends to be of an intermediate
form (>25 to 50 µmol/L). Little or no homocysteine is excreted by the
normal kidney. The role of the kidney in homocysteine metabolism and the
regulation of homocysteine metabolism is poorly understood. The homocystinurias,
rare inborn errors of homocysteine metabolism, are associated with severe
hyperhomocysteinemia (100 to 500 µmol/L) and premature atherosclerosis
and thrombosis.
How
does homocysteine injure blood vessels?
Because homocysteine is a thiol, it can undergo autooxidation and oxidation
with other thiols. The resulting reactive oxygen species—hydrogen
peroxide and superoxide anion radical—generate oxidative stress.
The concentration of plasma total cysteine is 20 to 30 times higher than
that of plasma tHcy, yet cysteine, which also undergoes similar oxidative
reactions, is not usually considered a risk factor. If oxidative stress
is not the mechanism for homocysteine-induced vascular dysfunction, is
there perhaps another, more attractive hypothesis? Yes, and it is related
to direct molecular targeting by homocysteine. Recent evidence suggests
that homocysteine may limit the bioavailability of nitric oxide, resulting
in the impairment of flow-mediated vasodilatation. The limited bioavailability
of nitric oxide could be due to nitrosothiol formation with homocysteine.
Homocysteine may also target specific proteins and impair their activity
and function through disulfide bond formation. The decreased binding of
tissue plasminogen activator to homocysteine-modified annexin II is a
case in point and may explain, in part, the procoagulant activity of homocysteine.
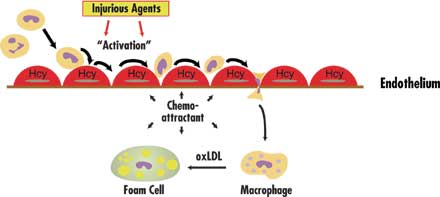
Finally, as shown in Figure 4, homocysteine may induce the expression
and secretion of chemokines such as monocyte chemoattractant protein 1
(MCP-1) and interleukin 8 (IL-8) in vascular endothelial cells. Production
of these chemokines by stimulated endothelial cells would attract monocytes
and neutrophils to sites of vascular injury where they could take up residence
in the intimal space.
|
|
| Figure 4. One
proposed mechanism for homocysteine (Hcy) involvement in vascular
disease. White blood cells (WBC) such as monocytes and neutrophils
flowing through blood vessels normally have random contact with vascular
endothelial cells (EC). When damage to ECs results from injurious
agents, however, these WBCs begin to roll along, and adhere to, the
endothelial surface. Homocysteine may speed the progression of vascular
disease by stimulating production of monocyte and neutrophil chemoattractants-MCP-1
and IL-8-in the vascular endothelium. Secretion is targeted to the
bottom side of the cell, thereby establishing a concentration gradient
for chemotaxis. Once attached, monocytes migrate between ECs and become
resident in the vascular intimal space. Here they are transformed
into macrophages, engulf oxidized low-density lipoprotein (LDL), and
become foam cells (the early observed lesion being called a fatty
streak). Foam cells are a source of reactive oxygen species which
can play a role in other sequences of events that promote atherosclerosis. |
Is
hyperhomocysteinemia a treatable disease?
Once a diagnosis of hyperhomocysteinemia has been made, it is safe and
easy to lower plasma tHcy in most individuals. A cocktail of folic acid
(400 to 800 µg), vitamin B12 (500 to 1000 µg) and vitamin B6 (25 to 100
mg) will reduce plasma tHcy up to 30 percent in subjects with cardiovascular
disease. Whether lowering homocysteine will have a beneficial effect on
disease progression will be known in 1 to 2 years after the completion
of a dozen or so worldwide clinical trials involving over 70,000 subjects.
Should
everyone be tested for plasma tHcy?
The American Heart Association has recommended that individuals with a
family history of heart and cardiovascular disease be tested for plasma
tHcy. Other subjects who should be tested are those with premature atherosclerosis
or atherosclerosis with no known conventional risk factors such as hypertension
or hyperlipidemia. Hypercoaguable profiles now routinely include plasma
tHcy. Of growing concern is the increased incidence of cognitive dysfunction
disorders, such as vascular dementia and Alzheimer's disease, and the
possibility that micronutrient deficiencies resulting in hyperhomocysteinemia
play a causative role. It may be common practice in the near future to
test everyone over the age of 60 for plasma tHcy.
Additional Reading
Hajjar KA, Mauri L, Jacovina AT, Zhong FM, Mirza UA, Padovan JC, et al.
Tissue plasminogen activator binding to the annexin II tail domain. Direct
modulation by homocysteine. J Biol Chem 1998;273:9987-93.
Jacobsen DW. Homocysteine
and vitamins in cardiovascular disease. Clin Chem 1998;44(8 Pt 2):1833-43.
Jacobsen DW. Hyperhomocysteinemia
and oxidative stress: time for a reality check? Arterioscler Thromb Vasc
Biol 2000;20:1182-4.
Lentz SR. Mechanisms
of thrombosis in hyperhomocysteinemia. Curr Opin Hematol 1998;5(5):343-9.
Mansoor MA, Svardal
AM, Ueland PM. Determination of the in vivo redox status of cysteine,
cysteinylglycine, homocysteine, and glutathione in human plasma. Anal
Biochem 1992;200(2):218-29.
Refsum H, Ueland P,
Nygård O, Vollset SE. Homocysteine and cardiovascular disease. Annu Rev
Med 1998;49:31-62.
Robinson K, Mayer
EL, Miller DP, Green R, van Lente F, Gupta A, et al. Hyperhomocysteinemia
and low pyridoxal phosphate. Common and independent reversible risk factors
for coronary artery disease. Circulation 1995;92:2825-30.
Robinson K, Gupta
A, Dennis V, Arheart K, Chaudhary D, . . . Jacobsen DW. Hyperhomocysteinemia
confers an independent increased risk of atherosclerosis in end-stage
renal disease and is closely linked to plasma folate and pyridoxine concentrations.
Circulation 1996;94:2743-8.
|